Hydrocarbon stapled peptides are mini-proteins locked into their bioactive alpha-helical conformation through site-specific introduction of a chemical brace.
The α-helix conformation being the most common element of protein secondary structures, has received considerable attention in drug discovery. High conformational instability of peptides leading to proteolytic cleavage and low bioavailability can be tackled via conformational stabilization of α-helical structure.
 Hydrocarbon stapled peptides are mini-proteins locked into their bioactive alpha-helical conformation through site-specific introduction of a chemical brace, an all-hydrocarbon staple. The idea of peptide stapling was introduced to overcome the limitations of two broad classes of therapeutic agents (small molecules and proteins-biologics) in targeting intracellular protein-protein interactions. As a rapidly emerging class of next-generation drugs stapled peptides are expected to combine the synthetic manipulability and cell-penetrating ability of small molecules with the three-dimensionality and versatile target recognition ability of biologics. Hydrocarbon stapled peptides are mini-proteins locked into their bioactive alpha-helical conformation through site-specific introduction of a chemical brace, an all-hydrocarbon staple. The idea of peptide stapling was introduced to overcome the limitations of two broad classes of therapeutic agents (small molecules and proteins-biologics) in targeting intracellular protein-protein interactions. As a rapidly emerging class of next-generation drugs stapled peptides are expected to combine the synthetic manipulability and cell-penetrating ability of small molecules with the three-dimensionality and versatile target recognition ability of biologics.
Two distinct conformational strategies are utilized to induce and stabilize α-helical structure, namely, α, α-di-substitution (helix nucleation by α-methylation) and macro-cyclic bridge formation (conformational constraint). Creation of a staple into a peptide thus entails the incorporation of two appropriately spaced, α-methyl, α-alkenylglycine residues, having defined stereo chemical configuration and alkene chain length, followed by ruthenium-mediated olefin metathesis on the synthesis resin and then release from the resin and deprotection to yield the stapled peptide. Three different types of all hydrocarbon staples are shown in the figure demonstrating the creation of stabilized α-helix in a peptide. The α-methyl, α-alkenylglycine amino acids utilized for the production of each staple type are indicated using the naming convention XY, where X is the stereochemistry at the a-carbon (Cahn–Ingold–Prelog designations) and Y is the length, in carbons, of the alkenyl side chain. During solid-phase peptide synthesis, α-Methyl, α-alkenylglycine cross-linking amino acids are incorporated in appropriate positions. An i, i+3 stapled peptide requires one unit of R5 at the i position and one unit of S5 at the i+3 position. An i, i+4 stapled peptide requires two units of S5 incorporated at the relative positions i and i+4. An i, i+7 stapled peptide requires one unit of R8 at the i position and one unit of S5 at the i+7 position. Resin-bound peptide is treated with Grubbs I olefin metathesis catalyst to produce a cross-link between the two non-natural amino acids, resulting in a stapled peptide that is braced in a α-helical conformation.
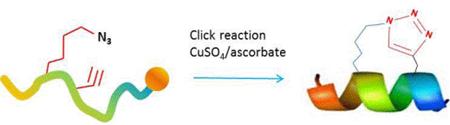 Stapled Peptides using Click Chemistry Stapled Peptides using Click Chemistry
The high efficiency and mild conditions of “click” reaction (Copper catalyzed Huisgen 1,3-dipolar cycloaddition reaction) combined with the ease of synthesis of the necessary unnatural amino acids allows for facile synthesis of triazole-stapled peptides. For example, a combination of L-Nle(εN3) and D-Pra (D-Propargylalanine) substituted at the i and i+4 positions can be used for the generation of single triazole-stapled peptides.
Modification of Stapled Peptides
Stapled peptide modifications typically fall into two categories: a fluorescent label or an affinity tag. Two of the most common moieties appended to the N-terminus of stapled peptides are fluorescein, which can be used for studies of intracellular uptake and biophysical characterization, and biotin, which can be used for biophysical characterization and assessment of in vitro target interaction. It is generally desired to include a flexible molecular spacer to isolate the modification from the core of the stapled peptide.
-
N-Acetylation
-
Linker attachment (β-alanine, mini-PEG etc.)
-
Fluorescent labeling (FITC, 5-FAM etc.)
Importance of stapling in drug design
The introduction of a hydrocarbon staple has been shown to confer high levels of α-helical content, and results in 5- to 5000-fold increase in target affinity, strong protection from proteolytic degradation, robust cell-penetration by endocytic vesicle trafficking, extension of in vivo half-life, and specific antagonism of protein–protein interactions in cultured cells.
-
Better target affinity
-
Increased proteolytic resistance and serum half-life
-
Cell penetration through endocytic vesicle trafficking
-
Target either extracellular or intracellular proteins
-
Disrupt Protein-Protein interactions
-
Non-immunogenic
-
Viable pharmacokinetics and in vivo stability
Stapled peptides have been studied in the targeting of several proteins which are relevant in Cancer, Diabetes, HIV, Atherosclerosis etc. These include proteins such as BCL-2, BCL-XL, BAX, MCL-1, glucokinase, hDM2, hDMX, NOTCH/CSL, HIV-1 capsid, HIV-1 gp41, ABCA1 and Estrogen receptor.
This paper describes the design of a series of novel stapled peptides that bind the co-activator peptide site of estrogen receptors. Using a number of biophysical techniques, including crystal structure analysis of receptor–stapled peptide complexes, the article describe in detail the molecular interactions and demonstrate that all-hydrocarbon staples modulate molecular recognition events. The findings have implications for the design of stapled peptides in general.
|

